前沿靶點速遞:每周醫學研究精選(十七)
日期:2024-10-30 11:42:17
靶點:TDO2
應用:非酒精性脂肪肝病
來源:Zhu Y, Shang L, Tang Y, et al. Genome-Wide Profiling of H3K27ac Identifies TDO2 as a Pivotal Therapeutic Target in Metabolic Associated Steatohepatitis Liver Disease. Adv Sci (Weinh). Published online October 4, 2024. doi:10.1002/advs.202404224
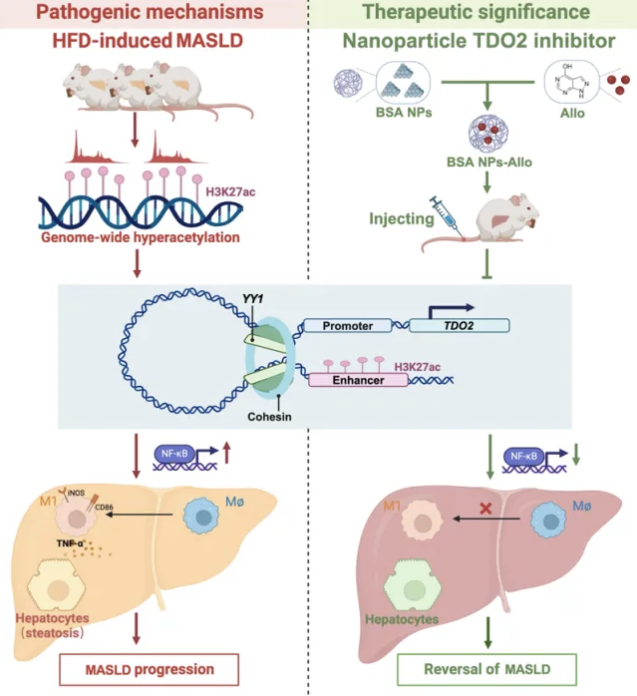
安徽醫科大學朱勇和朱亞玲研究團隊在《Advanced Science》期刊上發表的研究揭示了代謝功能障礙相關性脂肪肝病(MASLD,也稱為非酒精性脂肪肝病NAFLD)的表觀遺傳調控機制。研究通過多組學聯合分析,發現H3K27ac(組蛋白H3第27位賴氨酸的乙酰化)在轉錄因子YY1的調控下,能夠激活關鍵治療靶點TDO2,促進肝臟脂質積累和M1型巨噬細胞極化,加劇MASLD進展。研究還創新性地利用牛血清白蛋白納米粒子包裹阿洛普林(NPs-Allo)作為新型藥物遞送系統,有效緩解高脂飲食引起的代謝紊亂,為MASLD臨床治療提供新思路。這一發現不僅為MASLD的預防和治療提供了新視角,特別是在針對表觀遺傳標記物和關鍵分子途徑的干預方面,而且通過使用NPs-Allo納米粒子探索了潛在的治療方法,有效提高藥物療效并減少副作用,具有重要的科學和臨床意義。
靶點:FBXO3
應用:病毒研究
來源:Li H,Zhang Y,Rao G, et al. Rift Valley fever virus coordinates the assembly of a programmable E3 ligase to promote viral replication. Cell. 2024; doi:10.1016/j.cell.2024.09.008

《Cell》雜志上發表的研究中,科學家們發現了裂谷熱病毒(RVFV)非結構蛋白NSs通過重塑宿主E3泛素連接酶FBXO3來促進病毒復制的新機制。研究顯示,NSs蛋白能與FBXO3結合,形成NSs-FBXO3 E3連接酶,這一重塑的E3連接酶通過NSs-P62相互作用靶向轉錄因子IIH(TFIIH)復合物,導致TFIIH復合物降解,并阻斷抗病毒免疫應答。這項發現強調了FBXO3在RVFV感染中的關鍵作用,揭示了病毒如何利用宿主的泛素-蛋白酶體系統來降解關鍵的細胞蛋白,從而促進病毒的復制和發病機制。研究還發現,NSs纖維狀結構的形成對于體內病毒復制和病理過程至關重要,且NSs可以被改造成靶向其他蛋白質進行降解的多功能降解劑。這些發現為開發針對NSs的特異性抗RVFV藥物提供了新的方向,尤其是針對FBXO3的作用機制,為未來治療裂谷熱提供了新的策略。
靶點:SFTPC
應用:間質性肺病(ILD)研究
來源:Tang X, Wei W, Sun Y, et al. EMC3 regulates trafficking and pulmonary toxicity of the SFTPCI73T mutation associated with interstitial lung disease. J Clin Invest. Published online October 15, 2024. doi:10.1172/JCI173861

粵港澳大灣區精準醫學研究院(廣州)/復旦大學的唐曉芳團隊與Jeffrey A. Whitsett團隊合作,在《Journal of Clinical Investigation》上發表研究,揭示了內質網膜蛋白復合體關鍵亞基EMC3在調控肺表面活性蛋白C(SP-C)突變體SP-C(I73T)的轉運中的作用,并可作為間質性肺病(ILD)的潛在治療靶點。研究通過建立SftpcI73T敲入小鼠模型,發現SP-C(I73T)突變蛋白在AT2細胞內異常積累,導致細胞功能障礙。特異性敲除EMC3能改善肺部結構與功能,減少突變蛋白在細胞膜上的積累。研究人員還鑒定出EMC3的主要互作蛋白VCP,發現抑制EMC3表達或使用VCP抑制劑CB5083處理能改善突變蛋白的異常轉運和線粒體功能障礙。這項研究為理解SFTPCI73T突變導致ILD的機制提供了新見解,并為預防和治療相關疾病提供了新的靶點
靶點:Gαi2
應用:免疫調控研究
來源:Ham H,Jing H,Lamborn IT, et al. Germline mutations in a G protein identify signaling cross-talk in T cells. Science. 2024;385 (6715):eadd8947. doi:10.1126/science.add8947
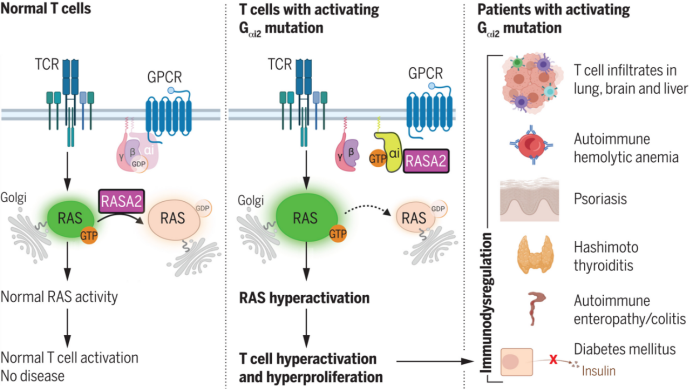
美國國立衛生研究院的Helen C. Su團隊在《Science》雜志上發表的研究揭示了G蛋白突變通過非經典途徑導致T細胞過度活化的新機制。研究團隊通過全外顯子組和全基因組測序識別出20名攜帶罕見或未報告的GNAI2基因突變的患者,這些突變導致Gαi2蛋白持續活躍,增強了對cAMP的抑制作用,影響免疫細胞的遷移能力。研究發現,突變的Gαi2蛋白通過RAS蛋白家族中的RASA2蛋白調節T細胞的活化和增殖,導致患者T細胞表現出異常的增殖反應,這一現象是通過RAS-MAPK和PI3K-AKT信號通路的增強導致的,而非通過經典的cAMP途徑。此外,GNAI2突變的影響不僅限于免疫系統,還涉及多個器官的發育缺陷。該研究定義了一種全新的免疫調控機制,解釋了Gαi2突變如何通過RAS信號通路引發T細胞過度活化,并為未來開發免疫相關疾病的治療策略提供了新思路。
靶點:ADCY7
應用:肝癌(HCC)研究
來源:Chen J,Jiang Y,Hou M, et al. Nuclear translocation of plasma membrane protein ADCY7 potentiates T cell-mediated antitumour immunity in HCC. Gut. 2024;:. doi:10.1136/gutjnl-2024-332902

海軍軍醫大學第三附屬醫院王紅陽院士和付靜教授領導的團隊在《Gut》雜志上發表的研究中發現了質膜蛋白ADCY7在肝癌(HCC)治療中的新機制。通過全基因組CRISPR篩選,研究者們揭示了ADCY7的核易位能夠顯著增強HCC中T細胞介導的抗腫瘤免疫反應。研究發現,在免疫功能正常的小鼠模型中,ADCY7的過表達能夠顯著抑制腫瘤生長,并且增加腫瘤中CD3+T細胞、CD8+T細胞以及IFN-γ+ CD8+T細胞的浸潤。進一步的分子機制探究顯示,ADCY7通過小窩蛋白介導的內吞作用進入細胞漿,并在LRRC59和KPNB1的協助下轉移到細胞核,作為CEBPA的共轉錄因子,促進CCL5的轉錄,從而增加CD8+T細胞的浸潤并抑制HCC的進展。此外,ADCY7還能被包裝進外泌體中,進一步促進CCL5的表達,吸引更多的CD8+T細胞。這一發現不僅闡明了ADCY7在HCC中調節T細胞免疫的機制,也為ADCY7作為增強免疫治療的潛在靶點提供了科學依據,有望改善HCC患者的免疫治療效果。
靶點:FABP5
應用:肝細胞癌研究
來源:Yang X,Deng B,Zhao W, et al. FABP5 + lipid-loaded macrophages process tumor-derived unsaturated fatty acid signal to suppress T-cell antitumor immunity. J Hepatol. 2024; doi:10.1016/j.jhep.2024.09.029
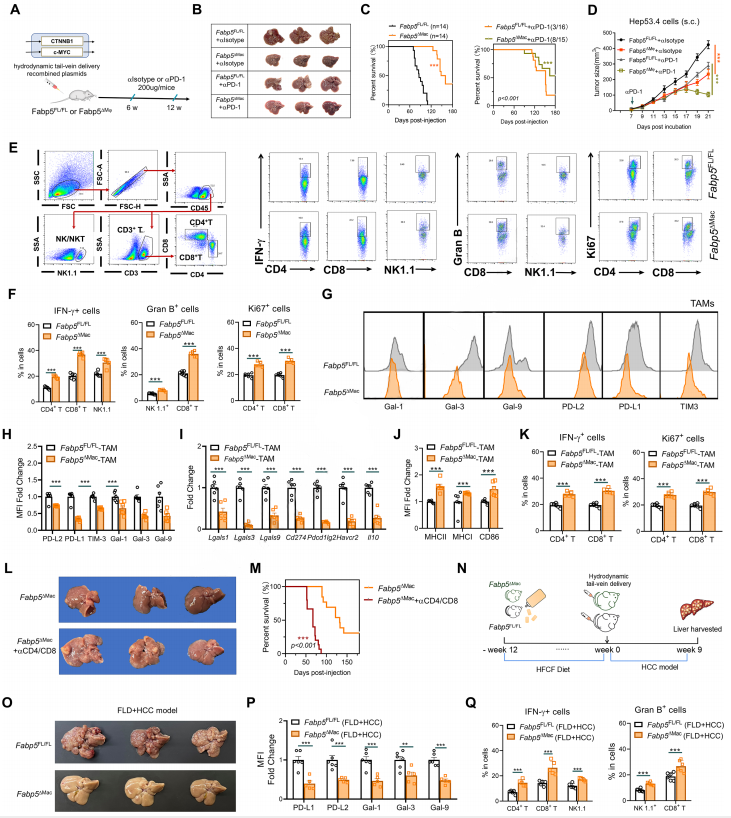
復旦大學上海醫學院、上海中醫藥大學附屬龍華醫院、復旦大學附屬中山醫院的林玉麗、楊旭光、黃曉武、徐延勇研究團隊在《Journal of Hepatology》上發表的研究揭示了FABP5+脂質負載巨噬細胞在肝細胞癌(HCC)中的作用。研究團隊通過單細胞RNA轉錄組測序技術,發現了一種具有顯著免疫抑制功能的FABP5+脂質負載巨噬細胞亞群。這些巨噬細胞能夠處理腫瘤來源的不飽和脂肪酸信號,表達多種免疫抑制分子,如GAL1、GAL3、GAL9、PD-L1及PD-L2,從而抑制T細胞的活化,削弱免疫治療的療效。這一過程依賴于FABP5與過氧化物酶體增殖物激活受體γ(PPARγ)的相互作用。研究還發現,通過靶向FABP5或干擾其功能可以削弱FABP5+巨噬細胞的免疫抑制表型,增強抗腫瘤T細胞的免疫反應,并延緩HCC的進展。這一發現不僅揭示了HCC腫瘤微環境中免疫抑制的新機制,也為開發針對FABP5+巨噬細胞的靶向免疫療法提供了理論基礎,為肝癌的免疫治療策略提供了新的方向。
靶點:Integrin-α5
應用:胰腺炎研究
來源:Gao RR, Ma LY, Chen JW, et al. ATN-161 alleviates caerulein-induced pancreatitis. J Genet Genomics. Published online October 11, 2024. doi:10.1016/j.jgg.2024.10.002
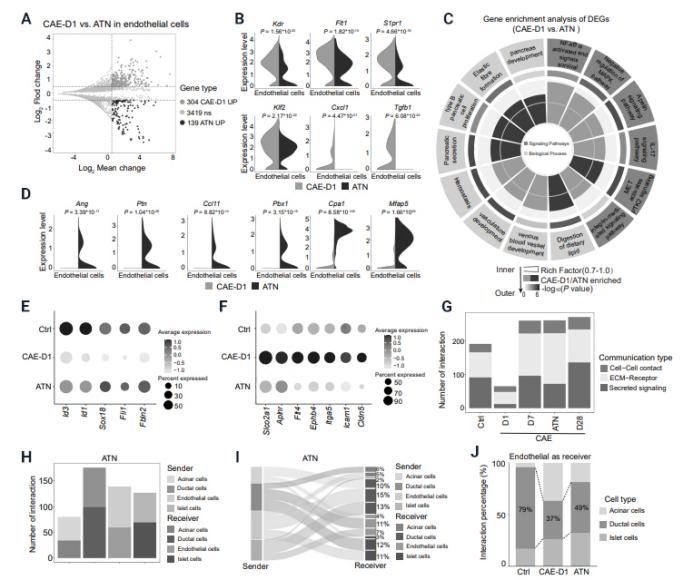
中國科學院廣州生物醫藥與健康研究院陳奇研究員團隊在《Journal of Genetics and Genomics》上發表的研究論文揭示了ATN-161通過靶向胰腺導管和血管細胞間的相互作用,有效緩解胰腺炎病理進程的新機制。研究通過雨蛙素誘導的胰腺炎動物模型和單細胞轉錄組測序分析,發現胰腺中導管上皮細胞與血管內皮細胞間存在緊密的并行關系,胰腺炎進程中伴隨異常的血管增生。研究指出,胰腺炎誘發CK19陽性細胞過量分泌Spp-1,通過內皮細胞上的受體Integrin-α5介導病理性血管新生。Integrin-α5的抑制劑ATN-161能顯著抑制胰腺炎中的腺泡-導管化生及病理性血管新生,并促進導管上皮細胞與血管內皮細胞間的正常互作。鑒于ATN-161在一期臨床實驗中顯示出良好的安全性,這一發現為胰腺炎治療提供了新的靶點和治療思路,具有重要的臨床應用潛力。
靶點:PAD4
應用:1型糖尿病(T1D)研究
來源:Shen Y, Shi R, Lu S, et al. Role of Peptidyl Arginine Deiminase 4-Dependent Macrophage Extracellular Trap Formation in Type 1 Diabetes Pathogenesis. Diabetes. 2024;73(11):1862-1874. doi:10.2337/db23-1000

中國藥科大學吳潔和樊竑冶團隊在《Diabetes》上發表的研究揭示了巨噬細胞胞外誘捕網(METs)在1型糖尿病(T1D)發病中的關鍵作用,特別是通過腸-胰軸的炎癥連接。研究指出,肽基精氨酸脫亞胺酶4(PAD4)是介導METs形成的關鍵酶,其活性敲除能減輕NOD小鼠的腸道炎癥,緩解T1D進程。PAD4敲除導致小鼠結腸中多個炎癥相關基因表達顯著下調,而胰腺中與蛋白折疊和β細胞再生相關基因顯著上調。此外,PAD4敲除降低了腸道中M1型巨噬細胞比例和METs水平,減少了腸系膜淋巴結和胰腺淋巴結中促炎性T細胞的比例。體外實驗驗證了PAD4在巨噬細胞向M1型極化和METs形成中的作用,而高糖環境會促進METs的形成。METs的過繼轉移實驗顯示,METs能增加小鼠促炎性T細胞比例,并促進T細胞的炎性分化和遷移。研究結果揭示了METs在加重腸道炎癥和通過腸-胰軸加速T1D疾病進程中的關鍵作用,為基于METs形成的T1D新型治療策略提供了實驗依據。
靶點:USP13
應用:肺腺癌研究
來源:Chen L, Ning J, Linghu L, et al. USP13 facilitates a ferroptosis-to-autophagy switch by activation of the NFE2L2/NRF2-SQSTM1/p62-KEAP1 axis dependent on the KRAS signaling pathway. Autophagy. Published online October 3, 2024. doi:10.1080/15548627.2024.2410619

中南大學基礎醫學院的陶永光研究員與張彬副教授在自噬期刊《Autophagy》上發表了研究論文,探討了肺腺癌細胞中鐵死亡與自噬之間的轉換機制。研究表明,去泛素化酶USP13通過激活NFE2L2/NRF2-SQSTM1/p62-KEAP1軸,依賴KRAS信號通路的激活,促進了鐵死亡向自噬的轉變。具體而言,USP13增加了NFE2L2的蛋白穩定性,進而增強了SQSTM1-LC3II通路介導的自噬,同時降低了SLC7A11的表達以抑制鐵死亡。這一發現揭示了KRAS突變型肺腺癌細胞在應對細胞應激時的復雜調控機制,強調了USP13在這一過程中扮演的重要角色。研究團隊認為,靶向USP13以促進自噬向鐵死亡的轉換,可能為治療KRAS突變型肺腺癌提供新的策略。該研究不僅為理解細胞死亡機制提供了新的視角,也為癌癥治療的潛在靶點開辟了新的方向。
參考文獻
1. Zhu Y, Shang L, Tang Y, et al. Genome-Wide Profiling of H3K27ac Identifies TDO2 as a Pivotal Therapeutic Target in Metabolic Associated Steatohepatitis Liver Disease. Adv Sci (Weinh). Published online October 4, 2024. doi:10.1002/advs.202404224
2. Li H, Zhang Y, Rao G, et al. Rift Valley fever virus coordinates the assembly of a programmable E3 ligase to promote viral replication. Cell. 2024; doi:10.1016/j.cell.2024.09.008.
3. Tang X, Wei W, Sun Y, et al. EMC3 regulates trafficking and pulmonary toxicity of the SFTPCI73T mutation associated with interstitial lung disease. J Clin Invest. Published online October 15, 2024. doi:10.1172/JCI173861.
4. Ham H, Jing H, Lamborn IT, et al. Germline mutations in a G protein identify signaling cross-talk in T cells. Science. 2024;385 (6715):eadd8947. doi:10.1126/science.add8947.
5. Chen J, Jiang Y, Hou M, et al. Nuclear translocation of plasma membrane protein ADCY7 potentiates T cell-mediated antitumour immunity in HCC. Gut. 2024;:. doi:10.1136/gutjnl-2024-332902.
6. Yang X, Deng B, Zhao W, et al. FABP5 + lipid-loaded macrophages process tumor-derived unsaturated fatty acid signal to suppress T-cell antitumor immunity. J Hepatol. 2024; doi:10.1016/j.jhep.2024.09.029.
7. Gao RR, Ma LY, Chen JW, et al. ATN-161 alleviates caerulein-induced pancreatitis. J Genet Genomics. Published online October 11, 2024. doi:10.1016/j.jgg.2024.10.002.
8. Shen Y, Shi R, Lu S, et al. Role of Peptidyl Arginine Deiminase 4-Dependent Macrophage Extracellular Trap Formation in Type 1 Diabetes Pathogenesis. Diabetes. 2024;73(11):1862-1874. doi:10.2337/db23-1000.
9. Chen L, Ning J, Linghu L, et al. USP13 facilitates a ferroptosis-to-autophagy switch by activation of the NFE2L2/NRF2-SQSTM1/p62-KEAP1 axis dependent on the KRAS signaling pathway. Autophagy. Published online October 3, 2024. doi:10.1080/15548627.2024.2410619.
應用:非酒精性脂肪肝病
來源:Zhu Y, Shang L, Tang Y, et al. Genome-Wide Profiling of H3K27ac Identifies TDO2 as a Pivotal Therapeutic Target in Metabolic Associated Steatohepatitis Liver Disease. Adv Sci (Weinh). Published online October 4, 2024. doi:10.1002/advs.202404224
(圖源:H3K27ac-TDO2-NF-κB軸在MASLD發展中發揮重要作用 [1])
安徽醫科大學朱勇和朱亞玲研究團隊在《Advanced Science》期刊上發表的研究揭示了代謝功能障礙相關性脂肪肝病(MASLD,也稱為非酒精性脂肪肝病NAFLD)的表觀遺傳調控機制。研究通過多組學聯合分析,發現H3K27ac(組蛋白H3第27位賴氨酸的乙酰化)在轉錄因子YY1的調控下,能夠激活關鍵治療靶點TDO2,促進肝臟脂質積累和M1型巨噬細胞極化,加劇MASLD進展。研究還創新性地利用牛血清白蛋白納米粒子包裹阿洛普林(NPs-Allo)作為新型藥物遞送系統,有效緩解高脂飲食引起的代謝紊亂,為MASLD臨床治療提供新思路。這一發現不僅為MASLD的預防和治療提供了新視角,特別是在針對表觀遺傳標記物和關鍵分子途徑的干預方面,而且通過使用NPs-Allo納米粒子探索了潛在的治療方法,有效提高藥物療效并減少副作用,具有重要的科學和臨床意義。
靶點:FBXO3
應用:病毒研究
來源:Li H,Zhang Y,Rao G, et al. Rift Valley fever virus coordinates the assembly of a programmable E3 ligase to promote viral replication. Cell. 2024; doi:10.1016/j.cell.2024.09.008
(圖源:doi:10.1016/j.cell.2024.09.008 [2])
《Cell》雜志上發表的研究中,科學家們發現了裂谷熱病毒(RVFV)非結構蛋白NSs通過重塑宿主E3泛素連接酶FBXO3來促進病毒復制的新機制。研究顯示,NSs蛋白能與FBXO3結合,形成NSs-FBXO3 E3連接酶,這一重塑的E3連接酶通過NSs-P62相互作用靶向轉錄因子IIH(TFIIH)復合物,導致TFIIH復合物降解,并阻斷抗病毒免疫應答。這項發現強調了FBXO3在RVFV感染中的關鍵作用,揭示了病毒如何利用宿主的泛素-蛋白酶體系統來降解關鍵的細胞蛋白,從而促進病毒的復制和發病機制。研究還發現,NSs纖維狀結構的形成對于體內病毒復制和病理過程至關重要,且NSs可以被改造成靶向其他蛋白質進行降解的多功能降解劑。這些發現為開發針對NSs的特異性抗RVFV藥物提供了新的方向,尤其是針對FBXO3的作用機制,為未來治療裂谷熱提供了新的策略。
靶點:SFTPC
應用:間質性肺病(ILD)研究
來源:Tang X, Wei W, Sun Y, et al. EMC3 regulates trafficking and pulmonary toxicity of the SFTPCI73T mutation associated with interstitial lung disease. J Clin Invest. Published online October 15, 2024. doi:10.1172/JCI173861
(圖源:RNA干擾介導的VCP基因沉默改變MLE-15和HEK293T細胞中proSP-C(I73T)的定位 [3])
粵港澳大灣區精準醫學研究院(廣州)/復旦大學的唐曉芳團隊與Jeffrey A. Whitsett團隊合作,在《Journal of Clinical Investigation》上發表研究,揭示了內質網膜蛋白復合體關鍵亞基EMC3在調控肺表面活性蛋白C(SP-C)突變體SP-C(I73T)的轉運中的作用,并可作為間質性肺病(ILD)的潛在治療靶點。研究通過建立SftpcI73T敲入小鼠模型,發現SP-C(I73T)突變蛋白在AT2細胞內異常積累,導致細胞功能障礙。特異性敲除EMC3能改善肺部結構與功能,減少突變蛋白在細胞膜上的積累。研究人員還鑒定出EMC3的主要互作蛋白VCP,發現抑制EMC3表達或使用VCP抑制劑CB5083處理能改善突變蛋白的異常轉運和線粒體功能障礙。這項研究為理解SFTPCI73T突變導致ILD的機制提供了新見解,并為預防和治療相關疾病提供了新的靶點
靶點:Gαi2
應用:免疫調控研究
來源:Ham H,Jing H,Lamborn IT, et al. Germline mutations in a G protein identify signaling cross-talk in T cells. Science. 2024;385 (6715):eadd8947. doi:10.1126/science.add8947
(圖源:激活的Gαi2繞過cAMP調節人類免疫反應 [4])
美國國立衛生研究院的Helen C. Su團隊在《Science》雜志上發表的研究揭示了G蛋白突變通過非經典途徑導致T細胞過度活化的新機制。研究團隊通過全外顯子組和全基因組測序識別出20名攜帶罕見或未報告的GNAI2基因突變的患者,這些突變導致Gαi2蛋白持續活躍,增強了對cAMP的抑制作用,影響免疫細胞的遷移能力。研究發現,突變的Gαi2蛋白通過RAS蛋白家族中的RASA2蛋白調節T細胞的活化和增殖,導致患者T細胞表現出異常的增殖反應,這一現象是通過RAS-MAPK和PI3K-AKT信號通路的增強導致的,而非通過經典的cAMP途徑。此外,GNAI2突變的影響不僅限于免疫系統,還涉及多個器官的發育缺陷。該研究定義了一種全新的免疫調控機制,解釋了Gαi2突變如何通過RAS信號通路引發T細胞過度活化,并為未來開發免疫相關疾病的治療策略提供了新思路。
靶點:ADCY7
應用:肝癌(HCC)研究
來源:Chen J,Jiang Y,Hou M, et al. Nuclear translocation of plasma membrane protein ADCY7 potentiates T cell-mediated antitumour immunity in HCC. Gut. 2024;:. doi:10.1136/gutjnl-2024-332902
(圖源:doi:10.1136/gutjnl-2024-332902 [5])
海軍軍醫大學第三附屬醫院王紅陽院士和付靜教授領導的團隊在《Gut》雜志上發表的研究中發現了質膜蛋白ADCY7在肝癌(HCC)治療中的新機制。通過全基因組CRISPR篩選,研究者們揭示了ADCY7的核易位能夠顯著增強HCC中T細胞介導的抗腫瘤免疫反應。研究發現,在免疫功能正常的小鼠模型中,ADCY7的過表達能夠顯著抑制腫瘤生長,并且增加腫瘤中CD3+T細胞、CD8+T細胞以及IFN-γ+ CD8+T細胞的浸潤。進一步的分子機制探究顯示,ADCY7通過小窩蛋白介導的內吞作用進入細胞漿,并在LRRC59和KPNB1的協助下轉移到細胞核,作為CEBPA的共轉錄因子,促進CCL5的轉錄,從而增加CD8+T細胞的浸潤并抑制HCC的進展。此外,ADCY7還能被包裝進外泌體中,進一步促進CCL5的表達,吸引更多的CD8+T細胞。這一發現不僅闡明了ADCY7在HCC中調節T細胞免疫的機制,也為ADCY7作為增強免疫治療的潛在靶點提供了科學依據,有望改善HCC患者的免疫治療效果。
靶點:FABP5
應用:肝細胞癌研究
來源:Yang X,Deng B,Zhao W, et al. FABP5 + lipid-loaded macrophages process tumor-derived unsaturated fatty acid signal to suppress T-cell antitumor immunity. J Hepatol. 2024; doi:10.1016/j.jhep.2024.09.029
(圖源:巨噬細胞中耗盡Fabp5抑制肝細胞癌進展并增強抗腫瘤免疫 [6])
復旦大學上海醫學院、上海中醫藥大學附屬龍華醫院、復旦大學附屬中山醫院的林玉麗、楊旭光、黃曉武、徐延勇研究團隊在《Journal of Hepatology》上發表的研究揭示了FABP5+脂質負載巨噬細胞在肝細胞癌(HCC)中的作用。研究團隊通過單細胞RNA轉錄組測序技術,發現了一種具有顯著免疫抑制功能的FABP5+脂質負載巨噬細胞亞群。這些巨噬細胞能夠處理腫瘤來源的不飽和脂肪酸信號,表達多種免疫抑制分子,如GAL1、GAL3、GAL9、PD-L1及PD-L2,從而抑制T細胞的活化,削弱免疫治療的療效。這一過程依賴于FABP5與過氧化物酶體增殖物激活受體γ(PPARγ)的相互作用。研究還發現,通過靶向FABP5或干擾其功能可以削弱FABP5+巨噬細胞的免疫抑制表型,增強抗腫瘤T細胞的免疫反應,并延緩HCC的進展。這一發現不僅揭示了HCC腫瘤微環境中免疫抑制的新機制,也為開發針對FABP5+巨噬細胞的靶向免疫療法提供了理論基礎,為肝癌的免疫治療策略提供了新的方向。
靶點:Integrin-α5
應用:胰腺炎研究
來源:Gao RR, Ma LY, Chen JW, et al. ATN-161 alleviates caerulein-induced pancreatitis. J Genet Genomics. Published online October 11, 2024. doi:10.1016/j.jgg.2024.10.002
(圖源:Integrin-α5拮抗劑ATN-161減輕急性胰腺炎的病理狀況 [7])
中國科學院廣州生物醫藥與健康研究院陳奇研究員團隊在《Journal of Genetics and Genomics》上發表的研究論文揭示了ATN-161通過靶向胰腺導管和血管細胞間的相互作用,有效緩解胰腺炎病理進程的新機制。研究通過雨蛙素誘導的胰腺炎動物模型和單細胞轉錄組測序分析,發現胰腺中導管上皮細胞與血管內皮細胞間存在緊密的并行關系,胰腺炎進程中伴隨異常的血管增生。研究指出,胰腺炎誘發CK19陽性細胞過量分泌Spp-1,通過內皮細胞上的受體Integrin-α5介導病理性血管新生。Integrin-α5的抑制劑ATN-161能顯著抑制胰腺炎中的腺泡-導管化生及病理性血管新生,并促進導管上皮細胞與血管內皮細胞間的正常互作。鑒于ATN-161在一期臨床實驗中顯示出良好的安全性,這一發現為胰腺炎治療提供了新的靶點和治療思路,具有重要的臨床應用潛力。
靶點:PAD4
應用:1型糖尿病(T1D)研究
來源:Shen Y, Shi R, Lu S, et al. Role of Peptidyl Arginine Deiminase 4-Dependent Macrophage Extracellular Trap Formation in Type 1 Diabetes Pathogenesis. Diabetes. 2024;73(11):1862-1874. doi:10.2337/db23-1000
(圖源:PAD4介導的METs形成與T1D疾病進程之間的相關聯 [8])
中國藥科大學吳潔和樊竑冶團隊在《Diabetes》上發表的研究揭示了巨噬細胞胞外誘捕網(METs)在1型糖尿病(T1D)發病中的關鍵作用,特別是通過腸-胰軸的炎癥連接。研究指出,肽基精氨酸脫亞胺酶4(PAD4)是介導METs形成的關鍵酶,其活性敲除能減輕NOD小鼠的腸道炎癥,緩解T1D進程。PAD4敲除導致小鼠結腸中多個炎癥相關基因表達顯著下調,而胰腺中與蛋白折疊和β細胞再生相關基因顯著上調。此外,PAD4敲除降低了腸道中M1型巨噬細胞比例和METs水平,減少了腸系膜淋巴結和胰腺淋巴結中促炎性T細胞的比例。體外實驗驗證了PAD4在巨噬細胞向M1型極化和METs形成中的作用,而高糖環境會促進METs的形成。METs的過繼轉移實驗顯示,METs能增加小鼠促炎性T細胞比例,并促進T細胞的炎性分化和遷移。研究結果揭示了METs在加重腸道炎癥和通過腸-胰軸加速T1D疾病進程中的關鍵作用,為基于METs形成的T1D新型治療策略提供了實驗依據。
靶點:USP13
應用:肺腺癌研究
來源:Chen L, Ning J, Linghu L, et al. USP13 facilitates a ferroptosis-to-autophagy switch by activation of the NFE2L2/NRF2-SQSTM1/p62-KEAP1 axis dependent on the KRAS signaling pathway. Autophagy. Published online October 3, 2024. doi:10.1080/15548627.2024.2410619
(圖源:doi:10.1080/15548627.2024.2410619 [9])
中南大學基礎醫學院的陶永光研究員與張彬副教授在自噬期刊《Autophagy》上發表了研究論文,探討了肺腺癌細胞中鐵死亡與自噬之間的轉換機制。研究表明,去泛素化酶USP13通過激活NFE2L2/NRF2-SQSTM1/p62-KEAP1軸,依賴KRAS信號通路的激活,促進了鐵死亡向自噬的轉變。具體而言,USP13增加了NFE2L2的蛋白穩定性,進而增強了SQSTM1-LC3II通路介導的自噬,同時降低了SLC7A11的表達以抑制鐵死亡。這一發現揭示了KRAS突變型肺腺癌細胞在應對細胞應激時的復雜調控機制,強調了USP13在這一過程中扮演的重要角色。研究團隊認為,靶向USP13以促進自噬向鐵死亡的轉換,可能為治療KRAS突變型肺腺癌提供新的策略。該研究不僅為理解細胞死亡機制提供了新的視角,也為癌癥治療的潛在靶點開辟了新的方向。
參考文獻
1. Zhu Y, Shang L, Tang Y, et al. Genome-Wide Profiling of H3K27ac Identifies TDO2 as a Pivotal Therapeutic Target in Metabolic Associated Steatohepatitis Liver Disease. Adv Sci (Weinh). Published online October 4, 2024. doi:10.1002/advs.202404224
2. Li H, Zhang Y, Rao G, et al. Rift Valley fever virus coordinates the assembly of a programmable E3 ligase to promote viral replication. Cell. 2024; doi:10.1016/j.cell.2024.09.008.
3. Tang X, Wei W, Sun Y, et al. EMC3 regulates trafficking and pulmonary toxicity of the SFTPCI73T mutation associated with interstitial lung disease. J Clin Invest. Published online October 15, 2024. doi:10.1172/JCI173861.
4. Ham H, Jing H, Lamborn IT, et al. Germline mutations in a G protein identify signaling cross-talk in T cells. Science. 2024;385 (6715):eadd8947. doi:10.1126/science.add8947.
5. Chen J, Jiang Y, Hou M, et al. Nuclear translocation of plasma membrane protein ADCY7 potentiates T cell-mediated antitumour immunity in HCC. Gut. 2024;:. doi:10.1136/gutjnl-2024-332902.
6. Yang X, Deng B, Zhao W, et al. FABP5 + lipid-loaded macrophages process tumor-derived unsaturated fatty acid signal to suppress T-cell antitumor immunity. J Hepatol. 2024; doi:10.1016/j.jhep.2024.09.029.
7. Gao RR, Ma LY, Chen JW, et al. ATN-161 alleviates caerulein-induced pancreatitis. J Genet Genomics. Published online October 11, 2024. doi:10.1016/j.jgg.2024.10.002.
8. Shen Y, Shi R, Lu S, et al. Role of Peptidyl Arginine Deiminase 4-Dependent Macrophage Extracellular Trap Formation in Type 1 Diabetes Pathogenesis. Diabetes. 2024;73(11):1862-1874. doi:10.2337/db23-1000.
9. Chen L, Ning J, Linghu L, et al. USP13 facilitates a ferroptosis-to-autophagy switch by activation of the NFE2L2/NRF2-SQSTM1/p62-KEAP1 axis dependent on the KRAS signaling pathway. Autophagy. Published online October 3, 2024. doi:10.1080/15548627.2024.2410619.











